
Hội Chứng DiGeorge Là Gì? Nguyên Nhân, Triệu Chứng Và Cách Điều Trị
- 1. Hội chứng DiGeorge là gì?
- 2. Cơ chế và nguyên nhân gây hội chứng DiGeorge
- 3. Triệu chứng của hội chứng DiGeorge
- 4. Biến chứng của hội chứng DiGeorge
- 5. Chẩn đoán hội chứng DiGeorge
- 6. Cách điều trị hội chứng DiGeorge
- 7. Tiên lượng và khả năng sống của bệnh nhân DiGeorge
- 8. Cách phòng ngừa hội chứng DiGeorge
- 9. Khi nào nên đi khám bác sĩ?
- 10. Câu hỏi thường gặp về hội chứng DiGeorge
- Tổng kết
Trong số các rối loạn di truyền hiếm gặp ở trẻ em, hội chứng DiGeorge là một trong những tình trạng gây ảnh hưởng sâu rộng nhất đến sức khỏe tổng thể, đặc biệt là tim, hệ miễn dịch và phát triển trí tuệ. Hội chứng này có thể khiến trẻ mắc các dị tật tim bẩm sinh, dễ nhiễm trùng tái phát, hoặc gặp khó khăn trong học tập và giao tiếp. Tuy nhiên, nhờ những tiến bộ trong y học hiện nay, người mắc hội chứng DiGeorge vẫn có thể được chẩn đoán sớm và điều trị hiệu quả để sống khỏe mạnh.
Vậy hội chứng DiGeorge là gì, nguyên nhân do đâu, triệu chứng nhận biết ra sao, và điều trị như thế nào để kiểm soát bệnh tốt nhất? Bài viết dưới đây sẽ giúp bạn hiểu rõ toàn bộ về rối loạn di truyền này – từ cơ chế hình thành đến các hướng điều trị và chăm sóc khoa học nhất.
Tìm hiểu về hội chứng DiGeorge
1. Hội chứng DiGeorge là gì?
Hội chứng DiGeorge (tên tiếng Anh: DiGeorge Syndrome), còn được gọi là hội chứng mất đoạn 22q11.2, là một rối loạn di truyền hiếm gặp xảy ra khi một đoạn nhỏ của nhiễm sắc thể số 22 bị mất. Sự mất đoạn này làm ảnh hưởng đến sự phát triển của nhiều cơ quan trong cơ thể, bao gồm tim, tuyến ức, tuyến cận giáp và khuôn mặt.
Hội chứng được bác sĩ Angelo DiGeorge mô tả lần đầu tiên vào năm 1965, và hiện nay được xem là một trong những nguyên nhân phổ biến nhất của suy giảm miễn dịch bẩm sinh. Tần suất mắc ước tính khoảng 1 trên 4.000 trẻ sơ sinh, nhưng có thể cao hơn do nhiều trường hợp chưa được chẩn đoán.
2. Cơ chế và nguyên nhân gây hội chứng DiGeorge
Hội chứng DiGeorge xảy ra do mất đoạn nhỏ trên nhiễm sắc thể số 22 tại vị trí q11.2 (vì vậy còn gọi là hội chứng mất đoạn 22q11.2).
Đoạn gen này chứa hàng chục gen quan trọng cho sự hình thành và phát triển của tim, tuyến ức, tuyến cận giáp và cấu trúc sọ mặt. Khi bị mất, các cơ quan này không phát triển bình thường, dẫn đến hàng loạt bất thường lâm sàng.
2.1. Nguyên nhân di truyền
-
Khoảng 90% trường hợp là do đột biến mới (de novo) – nghĩa là không ai trong gia đình mang gen này, nhưng đột biến xảy ra ngẫu nhiên khi phôi thai hình thành.
-
Khoảng 10% còn lại có tính di truyền trội (autosomal dominant) – nghĩa là nếu cha hoặc mẹ mang đột biến, có 50% khả năng truyền cho con.
2.2. Các gen liên quan
Một trong những gen quan trọng nhất bị mất là TBX1, gen có vai trò trong sự phát triển của tim và mặt. Thiếu gen này là nguyên nhân chính gây dị tật tim và bất thường sọ mặt đặc trưng của hội chứng DiGeorge.
.webp)
Nguyên nhân gây ra hội chứng DiGeorge
3. Triệu chứng của hội chứng DiGeorge
Các biểu hiện của hội chứng DiGeorge rất đa dạng, không phải bệnh nhân nào cũng có đầy đủ các triệu chứng. Tuy nhiên, có một số nhóm biểu hiện chính thường gặp:
3.1. Dị tật tim bẩm sinh
Khoảng 75–80% trẻ mắc hội chứng DiGeorge có bất thường tim, bao gồm:
-
Thông liên thất (VSD) – lỗ hở giữa hai buồng tim dưới.
-
Tứ chứng Fallot (Tetralogy of Fallot) – gồm bốn dị tật kết hợp gây tím tái.
-
Gián đoạn cung động mạch chủ (Interrupted aortic arch).
-
Thân chung động mạch (Truncus arteriosus) – động mạch chủ và động mạch phổi không tách biệt.
Những bất thường này có thể gây tim bẩm sinh tím tái, suy tim, khó thở và chậm phát triển thể chất.
3.2. Suy giảm miễn dịch
Do tuyến ức kém phát triển hoặc không có, cơ thể không sản xuất đủ tế bào T (T-lymphocytes) – thành phần quan trọng trong hệ miễn dịch.
-
Trẻ dễ nhiễm trùng tái phát, đặc biệt là nhiễm virus và nấm.
-
Trong trường hợp nặng, có thể cần ghép tuyến ức hoặc ghép tủy xương để hồi phục miễn dịch.
3.3. Giảm canxi máu (do thiểu năng tuyến cận giáp)
-
Tuyến cận giáp không hoạt động bình thường dẫn đến hạ canxi máu, gây co giật, co thắt cơ, run rẩy.
-
Biểu hiện thường xuất hiện ngay sau sinh hoặc trong vài tuần đầu đời.
3.4. Đặc điểm khuôn mặt đặc trưng
Trẻ mắc hội chứng DiGeorge có thể có một số đặc điểm nhận dạng như:
-
Mắt xếch và xa nhau.
-
Sống mũi dẹt, mũi nhỏ.
-
Tai thấp hoặc hình dạng bất thường.
-
Cằm nhỏ (micrognathia).
3.5. Chậm phát triển và rối loạn học tập
-
Trẻ có thể chậm nói, chậm đi, hoặc gặp khó khăn trong học tập.
-
Một số trẻ có rối loạn phổ tự kỷ (ASD) hoặc rối loạn tăng động giảm chú ý (ADHD).
3.6. Rối loạn tâm thần và hành vi (ở người lớn)
Người trưởng thành có thể phát triển:
-
Rối loạn lo âu, trầm cảm, rối loạn lưỡng cực.
-
Nguy cơ mắc tâm thần phân liệt cao hơn 25 lần so với dân số bình thường.
.webp)
Triệu chứng của hội chứng DiGeorge
4. Biến chứng của hội chứng DiGeorge
Tùy mức độ nặng, hội chứng DiGeorge có thể dẫn đến nhiều biến chứng đe dọa sức khỏe:
-
Suy tim bẩm sinh do dị tật tim nặng.
-
Nhiễm trùng mạn tính do hệ miễn dịch yếu.
-
Co giật do hạ canxi máu.
-
Chậm phát triển trí tuệ và thể chất.
-
Vấn đề về tâm thần và hành vi.
-
Khó khăn trong học tập và hòa nhập xã hội.
Nhờ tiến bộ y học, nhiều trẻ mắc hội chứng này vẫn có thể sống khỏe mạnh đến tuổi trưởng thành nếu được điều trị và theo dõi đúng cách.
5. Chẩn đoán hội chứng DiGeorge
Chẩn đoán hội chứng DiGeorge dựa trên biểu hiện lâm sàng và xét nghiệm di truyền học.
5.1. Xét nghiệm di truyền
-
Xét nghiệm FISH (Fluorescent In Situ Hybridization): xác định mất đoạn 22q11.2.
-
Xét nghiệm vi mảng nhiễm sắc thể (Chromosomal microarray): cho kết quả chi tiết hơn, phát hiện cả mất đoạn nhỏ.
-
Xét nghiệm di truyền tiền sản: có thể phát hiện sớm trong thai kỳ nếu cha hoặc mẹ mang gen bệnh.
5.2. Xét nghiệm bổ sung
-
Siêu âm tim, điện tâm đồ (ECG): phát hiện dị tật tim.
-
Xét nghiệm máu đo nồng độ canxi và hormone tuyến cận giáp (PTH).
-
Đánh giá miễn dịch học (đếm tế bào T) để xác định mức độ suy giảm miễn dịch.
-
Đánh giá phát triển tâm thần – vận động để hỗ trợ can thiệp sớm.
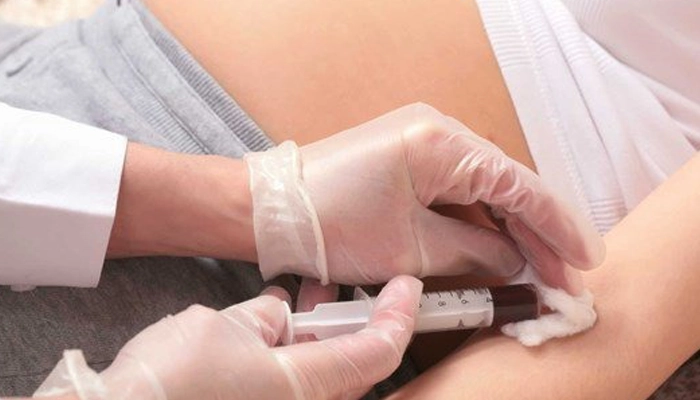
Chẩn đoán hội chứng DiGeorge dựa trên biểu hiện lâm sàng đa cơ quan và được xác định bằng xét nghiệm di truyền phát hiện mất đoạn 22q11.2.
6. Cách điều trị hội chứng DiGeorge
Hiện nay, không có phương pháp điều trị dứt điểm cho hội chứng DiGeorge vì đây là bệnh do mất đoạn nhiễm sắc thể. Tuy nhiên, điều trị triệu chứng và hỗ trợ đa chuyên khoa giúp cải thiện đáng kể chất lượng cuộc sống cho bệnh nhân.
6.1. Điều trị dị tật tim
-
Phẫu thuật tim bẩm sinh là biện pháp chính giúp sửa chữa cấu trúc tim.
-
Sau phẫu thuật, cần theo dõi lâu dài để kiểm soát huyết áp và chức năng tim.
6.2. Điều trị rối loạn miễn dịch
-
Với trường hợp suy miễn dịch nhẹ, có thể chỉ cần theo dõi và điều trị dự phòng nhiễm trùng.
-
Trường hợp nặng cần:
-
Ghép tuyến ức hoặc ghép tủy xương để tái tạo tế bào T.
-
Tránh tiêm vaccine sống (như sởi – quai bị – rubella, thủy đậu) vì có thể gây nhiễm bệnh.
-
6.3. Điều trị hạ canxi máu
-
Dùng canxi và vitamin D bổ sung lâu dài.
-
Theo dõi thường xuyên nồng độ canxi và phospho trong máu.
6.4. Hỗ trợ phát triển và tâm lý
-
Vật lý trị liệu, ngôn ngữ trị liệu và giáo dục đặc biệt giúp trẻ cải thiện khả năng học tập và giao tiếp.
-
Trị liệu tâm lý và thuốc an thần nhẹ có thể được chỉ định nếu có rối loạn cảm xúc hoặc hành vi.
6.5. Theo dõi định kỳ
Bệnh nhân nên được theo dõi định kỳ suốt đời bởi đội ngũ đa chuyên khoa gồm:
-
Bác sĩ tim mạch.
-
Bác sĩ nội tiết.
-
Bác sĩ miễn dịch học.
-
Bác sĩ tâm thần hoặc tâm lý học.
7. Tiên lượng và khả năng sống của bệnh nhân DiGeorge
Tiên lượng của hội chứng DiGeorge phụ thuộc vào:
-
Mức độ dị tật tim.
-
Tình trạng suy miễn dịch.
-
Khả năng kiểm soát hạ canxi.
Những trường hợp nhẹ có thể sống khỏe mạnh và học tập bình thường, trong khi trường hợp nặng có thể đòi hỏi chăm sóc y tế suốt đời.
Nhờ sự tiến bộ của phẫu thuật tim, ghép tuyến ức và điều trị miễn dịch, tuổi thọ trung bình của bệnh nhân đã tăng đáng kể, nhiều người có thể sống đến tuổi trưởng thành và lập gia đình.
8. Cách phòng ngừa hội chứng DiGeorge
Do nguyên nhân chủ yếu là đột biến gen ngẫu nhiên, hội chứng DiGeorge không thể phòng ngừa hoàn toàn. Tuy nhiên, có thể giảm nguy cơ bằng:
-
Tư vấn di truyền trước sinh nếu trong gia đình có người mang đột biến 22q11.2.
-
Xét nghiệm sàng lọc thai nhi bằng NIPT hoặc chọc ối, giúp phát hiện sớm bất thường nhiễm sắc thể.
-
Chăm sóc thai kỳ đúng cách, tránh phơi nhiễm với thuốc hoặc chất độc hại trong giai đoạn sớm của thai kỳ.

Tư vấn di truyền giúp giảm thiểu nguy cơ sinh ra trẻ mắc hội chứng DiGeorge
9. Khi nào nên đi khám bác sĩ?
Cha mẹ nên đưa trẻ đến khám ngay khi có các dấu hiệu nghi ngờ:
-
Dị tật tim bẩm sinh.
-
Co giật, run rẩy, co thắt cơ ở trẻ sơ sinh.
-
Dễ nhiễm trùng tái phát.
-
Chậm nói, chậm phát triển vận động.
-
Đặc điểm khuôn mặt bất thường.
Chẩn đoán sớm giúp trẻ được can thiệp kịp thời, cải thiện sức khỏe và cơ hội hòa nhập cộng đồng.
10. Câu hỏi thường gặp về hội chứng DiGeorge
10.1. Hội chứng DiGeorge có chữa khỏi được không?
Hiện nay chưa có cách chữa khỏi hoàn toàn, nhưng điều trị triệu chứng và can thiệp sớm giúp người bệnh sống khỏe và phát triển gần như bình thường.
10.2. Hội chứng DiGeorge có di truyền không?
Khoảng 10% trường hợp có yếu tố di truyền từ cha hoặc mẹ, còn lại là đột biến mới xuất hiện ngẫu nhiên.
10.3. Trẻ mắc hội chứng DiGeorge có thể đi học bình thường không?
Nếu được hỗ trợ sớm về ngôn ngữ và học tập, nhiều trẻ có thể học hòa nhập và phát triển kỹ năng tốt.
10.4. Có thể phát hiện hội chứng DiGeorge khi mang thai không?
Có. Bác sĩ có thể thực hiện xét nghiệm NIPT, chọc ối hoặc FISH để phát hiện mất đoạn nhiễm sắc thể 22q11.2 trước sinh.
10.5. Người lớn mắc hội chứng DiGeorge có nguy cơ bệnh tâm thần không?
Có. Người lớn mắc hội chứng này có nguy cơ cao hơn mắc trầm cảm hoặc tâm thần phân liệt, vì vậy cần được theo dõi sức khỏe tâm lý thường xuyên.
Tổng kết
Hội chứng DiGeorge là rối loạn di truyền phức tạp, ảnh hưởng đến nhiều cơ quan trong cơ thể như tim, hệ miễn dịch, tuyến cận giáp và hệ thần kinh.
Mặc dù chưa có cách chữa khỏi hoàn toàn, nhưng với điều trị sớm và hỗ trợ đa chuyên khoa, bệnh nhân hoàn toàn có thể sống khỏe mạnh, học tập và hòa nhập xã hội bình thường.
Nhận biết sớm, theo dõi định kỳ và chăm sóc toàn diện chính là chìa khóa để cải thiện tiên lượng lâu dài cho người mắc hội chứng DiGeorge.
Số lần xem: 271


















